Quantum Chemistry and Normal
Modes of Vibration
Purpose:
The purpose of this experiment is to
learn about ab initio
quantum chemistry packages and normal modes of small polyatomic molecules.
Equipment
and Chemicals:
A PC
running Spartan and a printer.
Directions:
See the instructor for directions on
how to load and run Spartan.
Calculations:
You will perform an ab initio
3-21G calculation on a small polyatomic molecule.
Your molecule is:
The calculation you will perform may be used to illustrate
the connection between charge polarity and the shapes of the frontier molecular
orbitals. You
will also have the opportunity to caIculate a vibrational spectrum and animate the motions for a few
characteristic modes.
1.
Build your molecule.
Select New from the File menu and select the pieces from
the model kit to build your
molecule. Click on Minimize under
the Build menu. In the lower right hand corner you will see
information about the energy of the minimum geometry of the molecule. Just below this, you will see the symmetry of
your molecule.
My molecule has ________________ symmetry.
Select View from
the Build menu to remove the model kit from the screen.
2. Enter the Calculations dialog (Setup menu). Under Calculate
specify Equilibrium Geometry with Hartree-Fock
with 3-21G(*)
for the level. Make certain that Total Charge
and Multiplicity are properly set (to
Neutral and Singlet, respectively), and select Elect Charges and Atomic
Charges. Then, click on OK to exit the
dialog.
3. Submit the job (Submit under the Setup menu). You will be asked to save the file and to supply a
name (e.g., “formaldehyde_321g”). When
complete examine the calculated charges on all of the atoms in the molecule. To
do this, first select an atom in the molecule by clicking on it and then click Properties from the Display menu. In the table below, record the Charges from the Atom Properties sub-menu which appears. Click on another atom in your molecule and
record the charges for that atom.
Continue until you have recorded the Electrostatic, Mulliken
and Natural charges for all atoms in your molecule.
Note that the Electrostatic, Mulliken
and Natural atomic charges supplied are different. This reflects the fact that charge cannot be
uniquely defined.
The charges on the atoms in my molecule are:
|
Atom |
Electrostatic |
Mulliken |
Natural |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Focus on the sets of charges on two bonding atoms. Do they
confirm your expectations about the polarity of the bonds in the molecule? For example, in formaldehyde, the
carbon-oxygen double bond, i.e.,
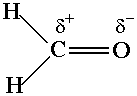
is polar with a partial
positive charge (d+) on C,
and a partial negative charge (d-) on O.
When you finish examining atomic charges, close the Atom Properties window and click on the
last selected atom to remove the high-lighting.
To display the dipole moment vector, select Properties from the Display menu and then Dipole from the sub-menu which appears. Draw a picture of the molecule with the
dipole vector:
Are the atomic charges consistent with the direction of the
dipole moment in your molecule? Yes or No?
Click on Dipole
to remove the dipole and close the Molecule
Properties window when you are finished.
4. Enter the Surfaces dialog (Setup menu). Click Add … to reach the sub-menu. Select HOMO
from the Surface pull-down menu then
Click OK. Next, click Add … and select LUMO from the Surface
menu and again click on OK. Close the window and submit the job (Submit from the Setup menu).
5. When the
calculations have completed, select Surfaces
from the Display menu. Click on the line "HOMO". Click
on the molecule and select Solid or
one of the other choices from among the available display styles on the bottom
right of the screen (the Style: pull-down menu). This will display the
Highest Occupied Molecular Orbital (HOMO) on the screen. Print
it out and attach to this lab report.
6. Click on the line “HOMO” to de-select the HOMO surface. This turns off the display of the HOMO. Now, Click on the line "LUMO". Once again, select Solid (or Mesh,etc.)
from among the available display styles.
This will display the Lowest Unoccupied Molecular Orbital (LUMO) of your
molecule. Print it out and attach to
this lab report. It is generally the
case that high-energy filled molecular orbitals
corresponding to polar bonds are polarized toward the more electronegative
atom, while low-energy unfilled molecular orbitals
are polarized toward the more electropositive atom.
When you are finished, turn off the display of the LUMO by clicking on the line "LUMO" and
close the SURFACES window.
8. Reenter the Calculations dialog (Set-up menu) and specify Freq. and Vibrational Modes. Exit the dialog (OK)
and then submit the job (Submit
under the Setup menu).
9. When the
calculation has completed select Vibrations
under the Display menu.
A dialog provides a listing of the vibrational
frequencies of your molecule (from smallest to largest) along with their
symmetry labels (e.g., A1 or B2). Record
these in the following table.
|
Symmetry
Label |
Calculated
Frequency |
Experimental
Frequency |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, that the calculated frequencies are consistently
larger than the measured values, typically by about 10%. It is common practice to uniformly scale calculated
frequencies by 0.9) to bring them into better accord with experiment.
To animate the individual vibrational
motions, click on the frequency in Vibrations dialog. You should be able to see whether or not the
motions you observe are consistent with the experimental assignments.
Attempt to sketch one of the normal modes of the
molecule. The following is an example of
this type of drawing for one of the normal modes of water:

10. Close the
“Vibrations” menu and remove your molecule from the screen by selecting Close from the File menu. And Exit from the File menu to close the program.